
Mặc dù khẳng định bám sát quy trình chẩn đoán theo khuyến cáo của Tổ chức Y tế Thế giới (WHO), Song, Viện Pasteur TP Hồ Chí Minh lại sử dụng kit Lightmix 1 “thành phần” (modular) không hề được WHO, FDA, EU, hay Bộ Y tế Việt Nam cấp phép để xét nghiệm Covid-19.
Như Tieudung.vn đã thông tin trước đó, trong gói thầu ký hợp đồng vào ngày 29/12/2020, Viện Pasteur đã mua bộ Lightmix có gene RdRp, trong khi gene RdRp đã bị WHO hướng dẫn loại bỏ trong xét nghiệm SAR-CoV-2. Và hơn thế, Lightmix là sản phẩm chưa có tên trong danh mục kit xét nghiệm Covid-19 mà Bộ Y tế cấp phép.
Liên quan đến nội dung này, ngày 1/4, Dược sĩ Hoàng Thuỳ Linh – Phó Giám đốc Trung tâm Kiểm định thiết bị y tế (Viện Pasteur TP Hồ Chí Minh) đã có văn phản hồi báo Kinh tế & Đô thị. Trong đó có 3 nội dung đáng chú ý như sau:
Nội dung thứ nhất, Viện Pasteur cho biết, sản phẩm Lightmix Modular phát hiện SARS-CoV-2 dựa trên khuếch đại gene E và khuếch đại gene RdRP (nhà sản xuất Tib Mobiol) được WHO thẩm định và khuyến cáo kể từ những ngày đầu dịch Covid-19 có độ nhạy đối với gen E là 5,2 RNA copies/μl, RdRp là 3,8 RNA copies/μl (Berlin protocol, WHO guidance: Tib Molbiol real-time RT-PCR assay for detection of Covid-19 virus).
“Là đơn vị phụ trách công tác phòng chống dịch phía Nam, với vai trò hướng dẫn, đánh giá chất lượng xét nghiệm Viện bám sát khuyến cáo của thế giới và tuân thủ quy định hiện hành của Việt Nam”, văn bản của Viện Pasteur nêu rõ.
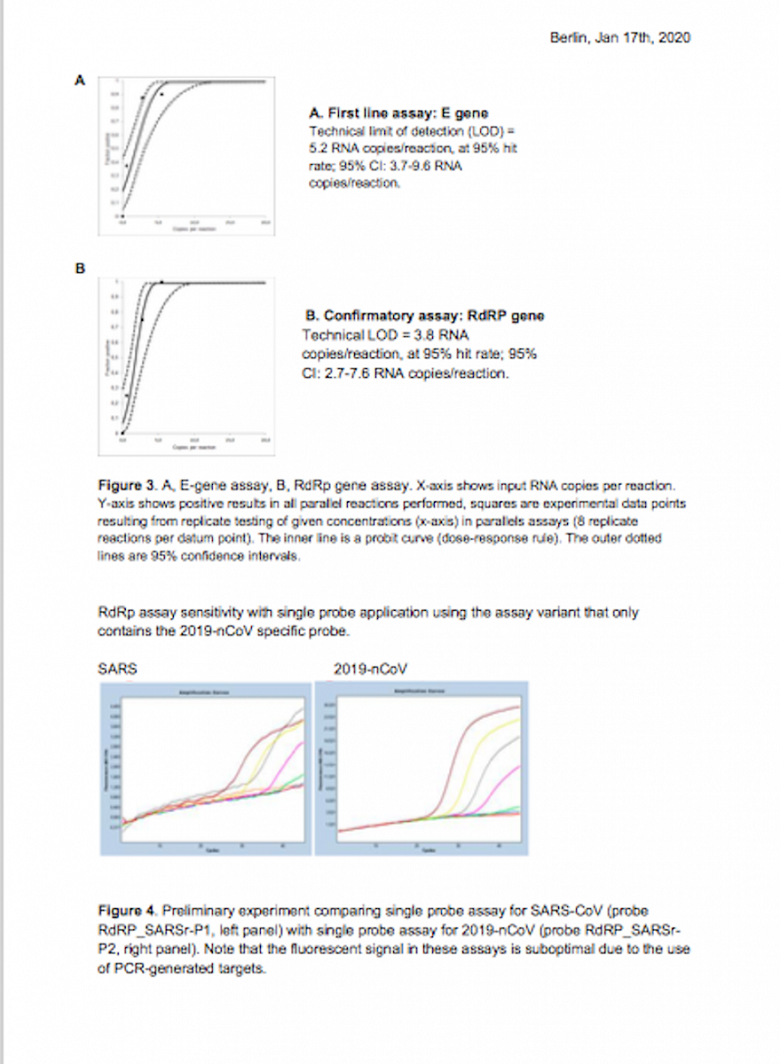
Tuy nhiên, theo tìm hiểu của PV, đúng là ban đầu trong tài liệu của WHO vào 17/1/2020 version 2 thì độ nhạy đối với gen E là 5,2 RNA copies/reaction (phản ứng, tương ứng với 5μl chứ không phải là /μl), RdRp là 3,8 RNA copies/reaction, nghĩa là gen RdRp nhạy hơn gen E. Tuy nhiên vào cuối 7/2020 đại diện WHO (Ms. Orla Condell) đã có email cho các bên liên quan hướng dẫn bỏ gen RdRp và chỉ còn dùng gen E. Theo đó, lý do lớn nhất khiến WHO quyết định hướng dẫn bỏ gen RdRp và chỉ dùng gen E là vì gen RdRp kém nhạy hơn nếu dùng E để sàng lọc và RdRp để khẳng định nhiễm Covid thì khi gen E dương tính, gen RdRp hoàn toàn có khả năng âm tính. Điều này rất nguy hiểm, có thể dẫn đến kết quả âm tính giả.

Với một đại dịch nguy hiểm như Covid-19 thì “bắt lầm còn hơn bỏ sót”, nghĩa là thà dương tính giả còn hơn âm tính giả. Chỉ cần một ca âm tính giả lọt ra cộng đồng, thì hậu quả sẽ không thể tưởng tượng được. Nhận thức rõ nguy cơ này, nên ngày 21/09/2020, dựa trên hướng dẫn của WHO, Bộ Y tế đã ban hành quyết định 4042/QĐ-BYT hướng dẫn bỏ gen RdRp chỉ dùng gen E.
Như vậy, cả WHO và Bộ Y tế đều đã ban hành hướng dẫn bỏ gen RdRp, và nếu Pasteur chịu làm theo hướng dẫn chỉ làm gen E thì sẽ không tốn kém tiền mua hóa chất, sinh phẩm… liên quan đến gen RdRp. Vậy tại sao vẫn cố chấp sử dụng gen RdRp, là câu hỏi lớn mà Viện Pasteur TP Hồ Chí Minh chưa đưa ra được lời giải thích thỏa đáng với PV báo Kinh tế và Đô thị.
Nội dung thứ hai, Viện Paster khẳng định, ngay từ đầu vụ dịch đã bám sát quy trình chẩn đoán theo khuyến cáo của WHO: Sàng lọc: gen E; Nếu dương tính gen E thì thực hiện tiếp gen RdRp, nếu dương tính thì khẳng định ca dương tính SAS-CoV-2. Viện đã bám sát quy trình, sàng lọc gen E sử dụng đoạn mồi IDT. Với các trường hợp ca dương tính, các ca bất thường về kết quả của các phòng xét nghiệm khác, sàng lọc gen E, dung đoạn mồi Lightmix Gen E, nếu dương tính gen E thì thực hiện tiếp Lightmix Gen RdRp để khẳng định ca dương tính.
Song, thực tế, “Gen E sử dụng đoạn mồi IDT” và “đoạn mồi Lightmix Gen E” là 2 cái khác nhau. “Gen E sử dụng đoạn mồi IDT” là theo tài liệu của WHO ngày 17/01/2020 version 2 (https://www.who.int/docs/default-source/coronaviruse/protocol-v2-1.pdf). Tài liệu của WHO khuyến cáo quy trình của Tib Mobiol, không hề có thông tin nào liên quan đến Lightmix trong này. Còn “đoạn mồi Lightmix Gen E” là 1 dạng sản phẩm thương mại của Tib Mobiol. Lightmix bắt đầu có tại Việt Nam từ đầu 4/2020 (lúc đó Pasteur mới đưa vào dự trù theo CV số 644/TTr-PAS ngày 6/4/2020 và đến 29/12/2020 Viện Pasteur TP Hồ Chí Minh mới ra quyết định phê duyệt trúng thầu).
Vậy thì, như Viện Pasteur TP Hồ Chí Minh trình bày ở trên. mẫu ở Viện Pasteur sẽ “sử dụng đoạn mồi IDT” còn mẫu các “ca dương tính, các ca bất thường về kết quả của các phòng xét nghiệm khác” gửi về thì “dùng đoạn mồi Lightmix”. Tuy nhiên, Lightmix bắt đầu có tại Việt Nam từ đầu 4/2020 và đến 29/12/2020 Viện Pasteur TP Hồ Chí Minh mới ra quyết định phê duyệt trúng thầu. Điều này đồng nghĩa, trước thời điểm tháng 4/2020 chắc chắn phải “sử dụng đoạn mồi IDT” cho cả các mẫu của Viện và “của các phòng xét nghiệm khác” gửi về. Tức là, “sử dụng đoạn mồi IDT” theo quy trình WHO khuyến cáo như trên hoàn toàn đảm bảo mà không nhất thiết phải “dùng đoạn mồi Lightmix”.
Thực tế hiện nay, hầu hết các cơ sở y tế tại Việt Nam (kể cả Viện Vệ sinh Dịch tễ Trung Ương) đang “sử dụng đoạn mồi IDT” theo quy trình WHO khuyến cáo như trên chứ ít “dùng đoạn mồi Lightmix”. Nguyên nhân là vì 2 loại này là tương đương nhau nhưng “sử dụng đoạn mồi IDT” giá thành rẻ hơn rất nhiều so với “dùng đoạn mồi Lightmix” (như trong quyết định trúng thầu số 1006/QĐ-PAS ngày 29/12/2020 thì 1000 test “đoạn mồi IDT gen E” chỉ 21 triệu trong khi đó với chỉ 96 test “đoạn mồi Lightmix gen E” đã có giá 12.1 triệu).
Đến đây, không thể không đặt câu hỏi, tại sao với 1 thành phần có trong quy trình WHO khuyến cáo (bộ E Assay của IDT) với giá rất rẻ (21tr/1000 test) không được mua hoàn toàn mà phải chen bộ Light không được cấp phép với giá cao hơn gần 5 lần vào (12.1tr/96 test? Đây chỉ đơn thuần là sự lãng phí hay còn có khuất tất nào khác, có lẽ chỉ có Viện Pasteur TP Hồ Chí Minh mới có câu trả lời rõ nhất?
Nội dung thứ ba, Viện Pasteur cam kết tuân thủ theo quy định của Bộ Y tế. Theo đó, nếu xét nghiệm phát hiện nhiễm vi rút bằng kỹ thuật Realtime RT-PCR cho kết quả dương tính, mẫu bệnh phẩm phải được gửi tới phòng xét nghiệm khẳng định SARS-CoV-2 để thực hiện xét nghiệm khẳng định (theo qui trình và sinh phẩm khuyến cáo của WHO và/hoặc CDC Hoa Kỳ).
Sản phẩm Lightmix Modular phát hiện SARS-CoV-2 dựa trên khuếch đại gen E và khuếch đại gen RdRP (nhà sản xuất TIBMOLBIOL) đã được WHO và CDC tài trợ cho Việt Nam (đính kèm PO của 2 tổ chức). Sinh phẩm này không những được WHO khuyến cáo sử dụng, còn là chiến lược của WHO và CDC tài trợ giúp các nước có nguồn sinh phẩm chất lượng, tin cậy trong công tác phòng chống dịch.
Quyết định số 4042/BYT-DP ngày 21/09/2020 cập nhật quyết định 2245/BYT-DP về việc phê duyệt kế hoạch xét nghiệm phát hiện nhiễm SARS-CoV-2 trong giai đoạn dịch COVID-19 là văn bản pháp lý hiện hành hướng dẫn toàn bộ công tác xét nghiệm phòng chống dịch COVID-19. Tại III, mục 4 Sinh phẩm xét nghiệm “Sinh phẩm xét nghiệm đã được Bộ y tế cho phép hoặc WHO hoặc CDC Hoa Kỳ thẩm định và khuyến cáo”. Sản phẩm Lightmix Modular (nhà sản xuất Tib Mobiol) được Bộ Y tế cho phép và WHO khuyến cáo.
Với nội dung này một lần nữa, các lý luận của Viện Pasteur TP Hồ Chí Minh có dấu hiệu…chống chế!
Vì sao? Việc cấp phép chỉ thực hiện với 1 thành phẩm hoặc 1 quy trình. Thành phẩm thường được gọi là kit và có đầy đủ các thành phần để thực hiện xét nghiệm. Thành phẩm được cấp phép phải là thành phẩm đã được cấp chứng nhận IVD (In Vitro Diagnostic – Dùng cho chẩn đoán) bởi WHO, FDA Hoa Kỳ, EU hoặc Bộ Y tế nước sở tại.
Còn quy trình là 1 hướng dẫn gồm các hóa chất, sinh phẩm và các bước để thực hiện xét nghiệm, người sử dụng sẽ theo hướng dẫn đó để mua hóa chất, sinh phẩm về thực hiện xét nghiệm. Quy trình để được Bộ Y tế cấp phép thường phải là quy trình của WHO hoặc CDC Hoa Kỳ. Hiện nay, quy trình WHO hướng dẫn là quy trình của hãng Tib Mobiol (Berlin, Đức). Như vậy, quy trình WHO hay CDC Hoa Kỳ hướng dẫn hay 1 thành phẩm IVD đã được cấp phép bởi WHO, FDA Hoa Kỳ, EU hoặc Bộ Y tế nước sở tại khi vào Việt Nam vẫn phải thông qua các thủ tục cấp phép của Bộ Y tế thì mới được sử dụng.
Chưa kể, sản phẩm Lightmix của Tib Mobiol không phải là quy trình, cũng không phải là thành phẩm IVD. Như phản hồi của Viện Pasteur TP Hồ Chí Minh nêu ở trên, kit Lightmix chỉ là “đoạn mồi” tương tự như “đoạn mồi IDT”. Muốn thực hiện được xét nghiệm phải mua thêm sinh phẩm và vật tư khác. Ngay trong cái tên “Lightmix Modular” cũng đã thể hiện rõ, Lightmix không phải là 1 thành phẩm mà chỉ là 1 “thành phần” (modular) của 1 thành phẩm. Nghĩa là Bộ Y tế không cấp phép cho Lightmix là hoàn toàn đúng. Và một khi, Bộ Y tế không cấp phép thì không thể sử dụng cho chẩn đoán (In vitro diagnostic) được, nhất là với dịch bệnh nguy hiểm như Covid-19 thì càng phải chặt chẽ tuyệt đối về pháp lý.
Điều này thể hiện rất rõ trong quyết định số 4043/BYT-TB-CT, chỉ có 12 kit phát hiện SARS-CoV-2 nhập khẩu được Bộ Y tế cấp phép, trong đó không có sản phẩm nào có tên thương mại là Lightmix cũng như không có hãng sản xuất nào là Tib Mobiol. Thêm vào đó, trên trang web của WHO, với các bộ kit phát hiện SARS-CoV-2 được WHO cấp phép, PV cũng không ghi nhận thấy bộ kit nào có tên thương mại là Lightmix cũng như không có hãng sản xuất nào là Tib Mobiol.
Như vậy, có thể khẳng định, không hề có bất kỳ thông tin nào chứng minh sản phẩm kit Lightmix của Tib Mobiol đã được WHO, FDA Hoa Kỳ, CDC Hoa Kỳ, EU hay Bộ Y tế Việt Nam cấp phép. Do đó, là đơn vị đầu ngành, Viện Pasteur TP Hồ Chí Minh mua và sử dụng sản phẩm Lightmix sẽ tiềm ẩn nhiều nguy cơ rủi ro trong công cuộc chống dịch Covid-19 của cả nước?!
theo Tiến Đạt – Tiểu Thúy (Tieudung)
